Article Text

Statistics from Altmetric.com
Inhaled corticosteroids and β agonists are the most frequently prescribed drugs in the management of chronic asthma. Current guidelines emphasise their complementary role. Inhaled corticosteroids are the treatment of choice for all but the mildest of disease. Short acting β agonists are recommended for “as required” relief of asthma symptoms, whereas long acting agents are indicated as a supplement to anti-inflammatory therapy if breakthrough symptoms persist.1 Thus, co-prescribing is commonplace. However, despite dynamic interactions between endogenous glucocorticoids and catecholamines in vivo, it is only recently that interest in the possibility of drug interactions has developed. Two topical and clinically relevant questions arise. Firstly, do positive interactions occur, thus providing theoretical justification for current trends to use combination products incorporating a long acting β agonist and a corticosteroid? Secondly, do negative interactions occur which might explain the apparent paradox that, despite increasing use of the two therapies over the last 30 years, the overall burden of asthma morbidity in most western countries has continued to increase?2
Effects of glucocorticoids on β receptor function
Endogenous adrenal glucocorticoids have an important facilitatory effect on β receptor function in vivo. In animals adrenalectomy results in a generalised loss of responsiveness to catecholamines.3 ,4 Conversely, the presence of glucocorticoids enhances β receptor mediated responses: myocardial contractility, hepatic and voluntary muscle glucose metabolism, and bronchial smooth muscle relaxation have been shown to increase.4 These actions occur at physiological concentrations of glucocorticoid.
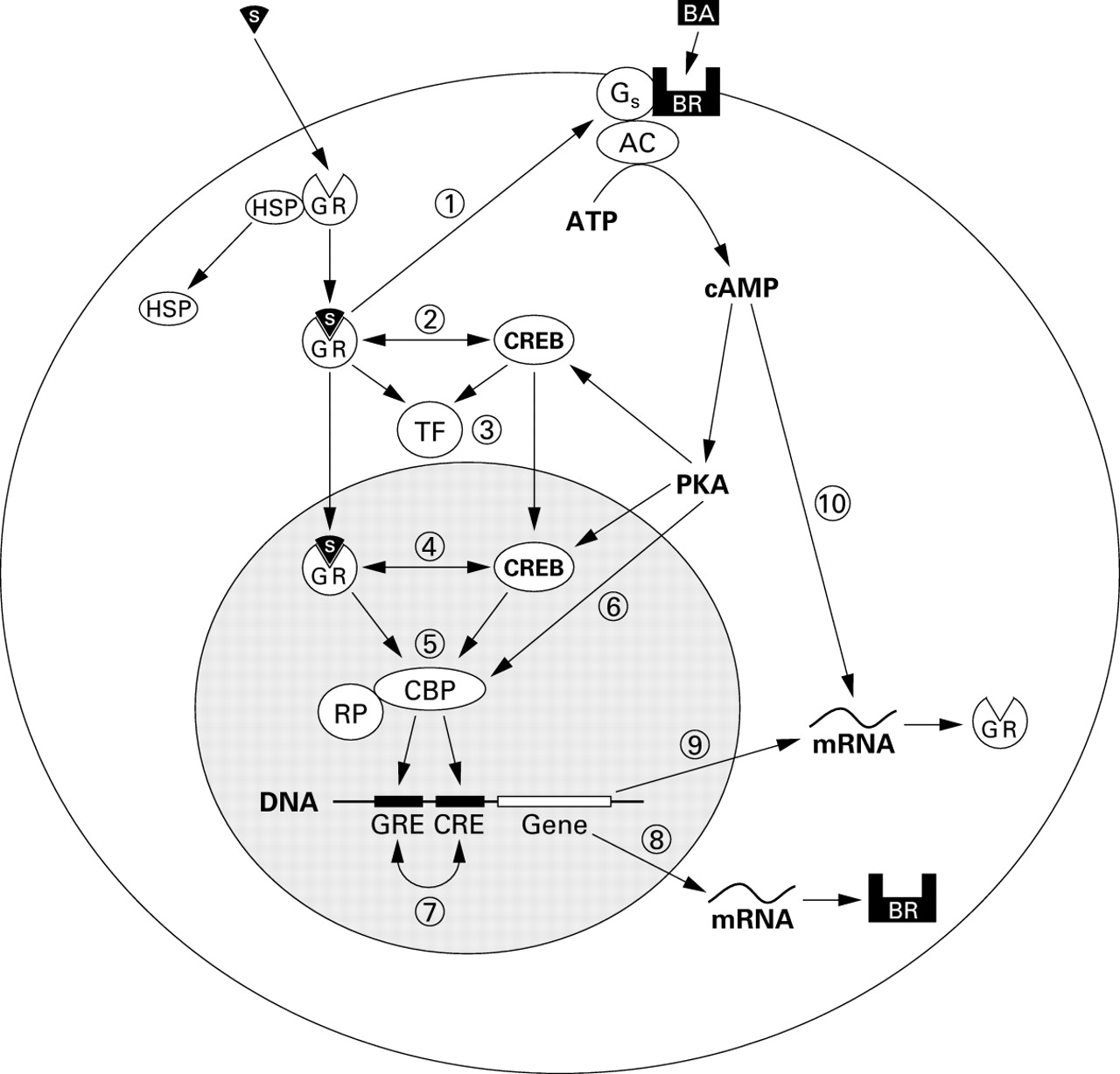
At least two mechanisms are proposed whereby glucocorticoids modify β receptor function. The first is by regulating the coupling of β receptors to G proteins and hence adenyl cyclase activation (fig 1). The degree of coupling determines cell responsiveness to β receptor stimulation.5 Following exposure to exogenous β agonist, uncoupling occurs rapidly by phosphorylation of the receptor (desensitisation). There are a number of phosphorylation pathways, including β adrenoceptor kinase (β-ARK),6 ,7 and these constitute the first step towards the development of drug tolerance. Exposure to corticosteroids restores receptors to their previously sensitised state.8
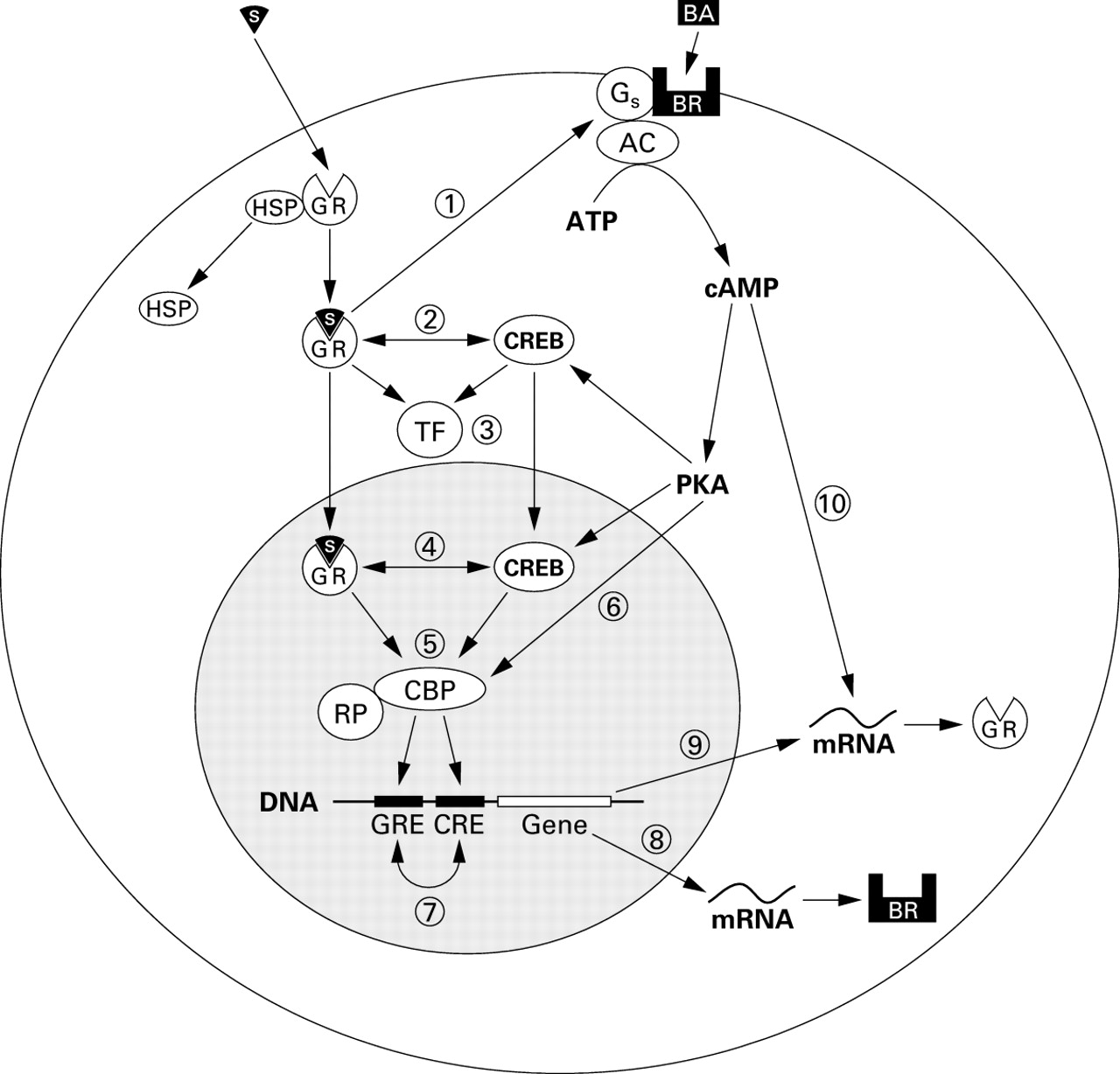
Putative intracellular mechanisms for interaction between β agonists and corticosteroids. (1) The steroid-glucocorticoid receptor complex upregulates β2receptors by enhancing β2 receptor coupling with G protein and/or preventing downregulation following β2receptor activation. (2) Binding between the activated glucocorticoid receptor and CREB in the cytoplasm may inhibit translocation to the nucleus. (3) “Cross talk” between the glucocorticoid receptor, CREB, and other nuclear transcription factors. (4) “Cross talk” between the glucocorticoid receptor and CREB in the nucleus. (5) The glucocorticoid receptor and CREB may compete for or synergistically bind protein co-factors such as CBP which are required for coupling with RNA polymerase and activation of response elements in the promoter regions of target genes. (6) Direct phosphorylation of protein co-factors such as CBP may alter their activity. (7) The presence of both GREs and CREs in the promoter regions of target genes may mutually enhance or inhibit their effects on gene transcription. (8) The promoter region of the β2 receptor gene contains GREs which may enhance or suppress synthesis of the β2receptor protein. (9) The promoter region of the glucocorticoid receptor gene contains CREs which may enhance or suppress synthesis of the glucocorticoid receptor protein. (10) A cAMP-dependent mechanism enhances the stability of messenger RNA for the glucocorticoid receptor increasing receptor levels.17 AC = adenylate cyclase; ATP = adenosine triphosphate; BA = β agonist; BR = β2adrenoceptor; CBP = CREB binding protein; CRE = cAMP response element; CREB = cAMP response element binding protein; cAMP = cyclic adenosine monophosphate; GR = glucocorticoid receptor; GRE = glucocorticoid response element; Gs = stimulatory G protein; HSP = heat shock protein 90; mRNA = messenger RNA; PKA = protein kinase A; RP = RNA polymerase; S = steroid drug molecule; TF = transcription factor.
The second is that glucocorticoids can upregulate previously downregulated β receptor function after chronic β agonist exposure. Downregulation is characterised by receptor internalisation and degradation,9 reversal of which requires new receptor synthesis. Activation of glucocorticoid response elements (GREs) in the promoter region of the β receptor gene causes an increase in the rate of gene transcription and hence of receptor numbers.10-15
Effects of β agonists on glucocorticoid receptor function
Inactive glucocorticoid receptors (GRs) are bound to protein complexes (including heat shock protein, HSP-90) in the cytosol. After ligand binding, activated GRs dissociate and translocate to the nucleus where they bind to glucocorticoid response elements (GREs) in the promoter region of target genes. The effect of catecholamines on the normal function of GRs and GREs has not been studied in detail. Forskolin, which increases intracellular cyclic AMP levels (thereby mimicking β2 receptor activation), has been shown to increase rat hepatoma cell GR numbers and potentiate the production of dexamethasone induced neurotensin from the rat hypothalamic cells.16 Forskolin may also antagonise the downregulation of GRs induced by dexamethasone.17 These actions provide indirect evidence that β agonists, which also enhance intracellular cAMP levels, might enhance GR function.
Glucocorticoids mediate many of their anti-inflammatory effects by either activating or repressing gene transcription of cytokines. In addition to the binding of activated GRs to GREs in the nucleus, this may also occur indirectly when activated GRs interact with pro-inflammatory nuclear transcription factors—for example, AP-1 or NF-kappa B (NF-κB)—to suppress their effects on the upregulation of pro-inflammatory cytokine production (fig 1).18 ,19 Beta agonists modify gene transcription by increasing intracellular levels of cAMP and activating the nuclear transcription factor cAMP response element binding protein (CREB). In turn this binds to cAMP response elements (CREs) on target genes. So-called “cross talk” between these transcription factors has been postulated as a mechanism for interactions between β agonist and corticosteroid drugs. It is likely that “cross talk” includes competitive binding of protein co-factors such as CREB binding protein (CBP) or the related P300 which are necessary for the activation of transcription factor response elements to GRs.20 Interactions between GREs and closely related CREs in the promoter region of genes may also be important.
There is evidence that the effects of β agonists and glucocorticoids on nuclear transcription factors are mutually inhibitory. Incubation of rat lung with salbutamol and fenoterol inhibits GR binding to GREs, while incubation with dexamethasone inhibits the binding of CREB to CREs.21 The extent of mutual inhibition is of the order of 40–50%. Similar effects have been described in the human lung.22 ,23 Despite this, in a clinical study designed to examine transcription factor changes in bronchial mucosa during treatment of mild asthma with terbutaline and budesonide singly and in combination, Hancox et al found no evidence of a negative interaction.24 Although DNA binding of the transcription factor NF-κB was reduced in bronchial mucosa and GR binding increased with regular inhaled budesonide, the simultaneous administration of regular terbutaline did not alter the binding activity of either.24
In other studies in human bronchial epithelial cells an inhibitory effect of terbutaline on GR binding responses to budesonide has been shown, but occurred only when the β agonist was administered simultaneously; the effects of prior exposure to corticosteroid were unaffected by the subsequent addition of terbutaline.25Thus, the timing of exposure to the two agents may be of relevance in determining possible interactions. In contrast, in another study both salbutamol and salmeterol have been shown to enhance the activation of GC receptors and their binding to nuclear GREs.26
The results of other laboratory studies designed to evaluate possible interactions between β agonists and glucocorticoids on inflammatory cells and their cytokines have also been inconsistent. Salmeterol has been shown to enhance the steroid induced inhibition of allergen activated monocytes27 and also appears to potentiate fluticasone induced apoptosis in activated human eosinophils (by a factor of 2–3).28 Similarly, in airway smooth muscle cells the inhibitory effect of dexamethasone on tumour necrosis factor (TNF)-α mediated interleukin-8 production was potentiated by simultaneous incubation with both salbutamol and salmeterol.29 In contrast, Seldon et al have studied the effect of salbutamol on dexamethasone induced inhibition of TNF-α and GM-CSF production from stimulated monocytes but, even at high concentrations of salbutamol, no important interaction was observed.30 Other in vitro data suggest that, at pharmacological concentrations, the presence of either isoprenaline or salbutamol may actually impair the beneficial actions of steroid. Nielson et al 31have shown that the effects of dexamethasone on eosinophil superoxide production and apoptosis are reduced in a dose dependent manner by both of these short acting β agonists.
A number of biological and pharmacological factors might account for the apparently conflicting outcomes from these studies. They include tissue dependent differences in receptor function, variations in the in vitro drug concentrations used and the duration of exposure, and differing pharmacological properties of the drugs used—for example, partial versus full agonist or short acting versus long acting. Nevertheless, they provide some of the background against which potential interactions between β agonists and inhaled corticosteroids in patients requiring chronic treatment have been investigated.
Clinical interactions: acute severe asthma
REVERSING DESENSITISATION AND DOWNREGULATION
Although the principal mode of action of corticosteroids in acute asthma is to control airway inflammation, several studies indicate that reversing β receptor desensitisation and downregulation caused by prior β agonist treatment may be important. In animal studies a bolus of corticosteroid reverses tolerance to the protective effects of isoprenaline against non-specific airway responsiveness.32However, at least in vitro, the magnitude of this effect appears to differ depending on which drugs are used.33 In humans, single doses of either intravenous prednisolone34 or even inhaled beclomethasone35 have been shown to restore the bronchodilator response to isoprenaline and fenoterol, respectively. Similarly, Tan et al 36 have shown that tolerance to the bronchodilator effects of inhaled formoterol may be rapidly reversed by a single dose of intravenous corticosteroid. The apparently short time frame for this change contrasts with that for the formation of cytokines and inflammatory cell infiltration in the late asthmatic response, suggesting that the initial benefits of steroid therapy in acute asthma are due to their permissive effect on β receptor function rather than suppression of pro-inflammatory pathways. This might also explain the clinical observation that, in acute severe asthma, the co-administration of corticosteroid either systemically or by the inhaled route results in an enhanced rate of recovery of lung function compared with β agonist or corticosteroid alone.37 ,38
Although interactions between β agonists and corticosteroids in acute asthma are likely to be beneficial, there are theoretical reasons why this might not always be the case. A number of hypotheses have been advanced to explain the relationship between the use of inhaled β agonists and the two recent “epidemics” of asthma mortality.39 Among them is the possibility that, in association with the hypoxia of acute severe asthma, overuse of β agonists might precipitate death by cardiac arrhythmia.40In patients who have been taking β agonists frequently or regularly, administering systemic corticosteroids might re-sensitise myocardial as well as bronchial smooth muscle β receptors, thus theoretically increasing the possibility of arrhythmias. In laboratory animals corticosteroid administration has been shown to increase the cardiac toxicity of catecholamines.41 Recently, Azizet al have shown that downregulation of human heart rate responses to salbutamol during treatment with the long acting β agonist formoterol was modified by concomitant use of inhaled steroid.42 Likewise, cardiovascular responses to emergency β agonist drugs may be increased in patients who have previously received treatment with inhaled corticosteroids.43 In contrast, in healthy volunteers the use of oral prednisone failed to potentiate the effects of β agonist on heart rate or QTc interval.44 Given that death from acute severe asthma is more likely to be the result of respiratory failure than cardiac causes,45 it seems unlikely that recovery or even enhancement of the downregulated cardiovascular effects of β agonist by corticosteroid during acute severe asthma is clinically important.
Clinical interactions: chronic asthma
PROTECTION AGAINST THE DEVELOPMENT OF TOLERANCE
If corticosteroids are of benefit in restoring β receptor numbers and function in acute asthma, the question arises: do they offer protection against the development of tolerance when administered as chronic therapy? Unfortunately, since no firm relationship has been established between β agonist tolerance during long term use of either short or long acting agents and asthma control, the clinical relevance of this question is speculative. Nevertheless, a number of studies have been carried out to establish whether inhaled corticosteroids may have a prophylactic role in preventing β agonist tolerance.
The results of most studies indicate that inhaled corticosteroids do not modify the development of tolerance during treatment with short acting agents.46-49 In a recent study by Hancoxet al 50 clear evidence of tolerance to the bronchodilator action of inhaled β agonist was seen in 34 asthmatic patients following six weeks treatment with regular inhaled terbutaline in a dose of 1 mg four times daily. After inducing bronchoconstriction with methacholine, the acute response to sequential doses of inhaled salbutamol was significantly reduced compared with placebo but the addition of regular budesonide to terbutaline in a dose of 400 μg twice daily did not prevent this effect. In one in vitro study some protection against the development of tolerance was achieved with the administration of dexamethasone,15 which is arguably a more potent corticosteroid. However, in a clinical study by Molema et al in which a range of doses of inhaled beclomethasone was given, tolerance to the acute effects of inhaled salbutamol remained unchanged.47 It therefore seems unlikely that the use of even high doses of inhaled corticosteroids will confer protection, even if avoiding tolerance were deemed to be clinically desirable.
A similar picture has emerged for the long acting β agonists, particularly with regard to the loss of protective effect against bronchoconstrictor stimuli afforded by single doses of a short acting drug during long term treatment with a long acting agent.51-56 In general, inhaled corticosteroids do not prevent the advent of tolerance, although here the evidence is perhaps less clear. Recently, Giannini et al have reported that inhaled beclomethasone partially reverses the loss of protective effect of salmeterol against allergen challenge.57 In another study with healthy volunteers Tanet al found that oral steroid prevented downregulation of systemic responses to salbutamol during long term treatment with formoterol.58
CLINICAL INTERACTIONS: SHORT ACTING β AGONISTS
In the late 1980s case control studies carried out in New Zealand identified an association between asthma mortality and the prescription of the β agonist fenoterol. The risk of death was greatest among those requiring continuous treatment with oral prednisone.59-61 This may not be surprising since these patients had more severe asthma, but it did raise the possibility of an adverse interaction. Further, the results of several investigations have found that airway inflammation may be increased during monotherapy with a short acting β agonist.62-64 Thus, individual case reports have suggested that reducing, or even withdrawing, short acting β agonist treatment may result in clinical improvement and a significant reduction in steroid requirements.65-67 These data have generated the hypothesis that the benefits of corticosteroid treatment may be compromised or, alternatively, that the need for anti-inflammatory therapy may be increased as a result of β agonist use and an adverse interaction between the two drugs.
Other clinical studies have offered support for this possibility. Wonget al 68 showed that the addition of regular terbutaline treatment resulted in significant impairment of the reduction in bronchial hyperresponsiveness to allergen resulting from inhaled budesonide (fig 2).

Changes in PC20 allergen (doubling doses) 33 hours after cessation of treatment with placebo, terbutaline, budesonide, and their combination. The increase in PC20which occurred with budesonide was significantly greater than for the combination of terbutaline and budesonide. Shaded areas = interquartile ranges. Reproduced from Wong et al68 with permission.
Similarly, in a controlled crossover study Cockcroftet al 69 showed that bronchial responsiveness to allergen was increased after treatment with regular salbutamol, reduced after treatment with budesonide, and intermediate when both drugs were used in combination.
Given this background, there has been a need for appropriately designed studies to confirm whether these observations are relevant to asthma control during long term combined therapy with inhaled corticosteroids and β agonists. A number of investigations have compared the addition of inhaled corticosteroids to β agonist treatment alone.70-75 Their results have been consistent: symptoms, pulmonary function, bronchial hyperresponsiveness, and exacerbation rates are improved using combined therapy. However, comparisons between combined treatment and inhaled corticosteroids alone, which are much more relevant to answering the question of a possible adverse interaction between the two drugs, have been fewer. Their results are summarised in table 1.63 ,76-81
Studies comparing short acting β agonists in combination with inhaled corticosteroids with inhaled corticosteroid alone
We have recently reported the results of two placebo controlled, four way, crossover studies designed to evaluate interactions between β agonists and inhaled corticosteroids.63 ,81 Neither investigation provided evidence that short acting β agonists reduced the clinical benefits of inhaled corticosteroids. In the larger study comprising 61 patients with mild to moderate asthma81combined treatment with inhaled terbutaline (1 mg four times daily) and inhaled budesonide (800 μg daily) over six weeks was ranked best of the four treatments, and achieved significant improvements in asthma control compared with budesonide alone. A similar positive interaction was noted for morning peak flows which showed a greater increase during combined treatment (compared with placebo) than the sum of the increases for terbutaline or budesonide given alone (fig 3) although, interestingly, this pattern was not true for FEV1. A simple explanation for these results is that terbutaline enhanced airway deposition of the budesonide and hence overall improvement. However, given the in vitro evidence already cited, a more complex interaction seems likely. The possibility that inhaled steroid may have offered protection against the development of tolerance has been investigated and discounted.50
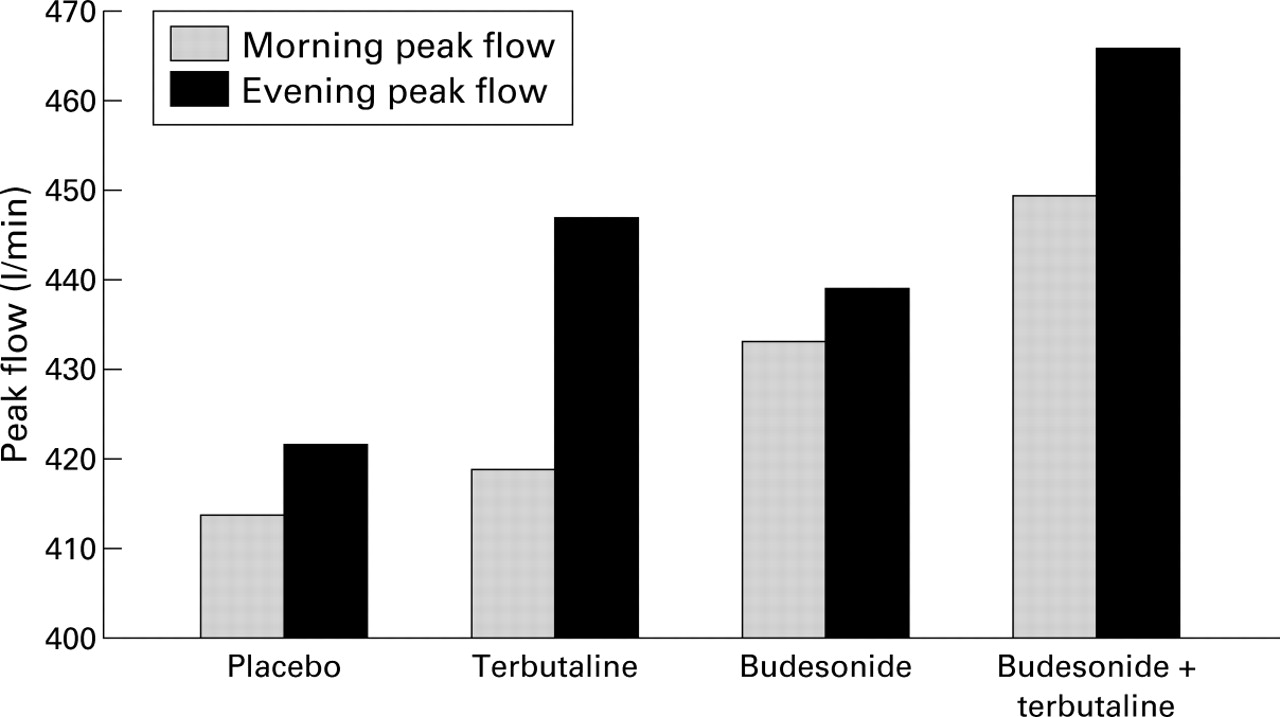
Mean morning and evening peak flow rates during six weeks treatment with regular inhaled terbutaline, budesonide, their combination, and placebo. Morning peak flows were higher when terbutaline was added to budesonide (p<0.002) whereas, when compared with placebo, terbutaline alone conferred no significant improvement. Reproduced from Hancox et al81 with permission.
Similar clinical outcomes were obtained in a second separate study comprising 34 patients.63 However, in that investigation there was a significant reduction in PD15 for hypertonic saline and a significant increase in the percentage of eosinophils in induced sputum during treatment with terbutaline, indicating that monotherapy with the short acting β agonist had a permissive effect on airway inflammation (fig 4). This adverse outcome was not modified by the addition of budesonide. Indeed, the pattern of results for these end points was similar to that previously described by Wonget al 68 and Cockcroftet al 69—namely, there was some impairment of the benefits of treatment with inhaled corticosteroids when the β agonist was added without apparently affecting asthma control.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes in (A) bronchial hyperresponsiveness to hypertonic saline (PD15 saline) and (B) percentage of eosinophils in induced sputum at the end of six weeks treatment with inhaled terbutaline (1 mg four times daily), budesonide (400 μg twice daily), combined treatment, and placebo given to 34 subjects in a randomised crossover sequence. Reproduced from Aldridge et al63 with permission.
Taken together, the results of these recent studies suggest that, even though inhaled corticosteroids do not appear to prevent the pro-inflammatory effects of short acting β agonists, this is unlikely to be of clinical significance, at least in patients with mild or moderate asthma taking “normal” doses of either drug. In fact, a beneficial interaction appears to occur in most patients. The results suggest that a β agonist facilitates the actions of inhaled corticosteroids, perhaps simply by improving airway drug deposition or, alternatively, by enhancing the anti-inflammatory effects of inhaled corticosteroids at a subcellular level.26-28
CLINICAL INTERACTIONS: LONG-ACTING β AGONISTS
Current guidelines for the management of chronic asthma support the addition of a long acting β agonist in patients whose asthma remains inadequately controlled with anti-inflammatory therapy and “as required” bronchodilator.1 As monotherapy, inhaled corticosteroid achieves greater control of asthma than long acting β agonist alone.82 ,83 However, the addition of a long acting agent results in greater improvements in symptoms and lung function than increasing the dose of inhaled corticosteroid.84-90
Although in some studies these outcomes do not appear to translate into a reduction in asthma exacerbations,91 ,92 this is perhaps because these studies were not primarily designed to evaluate this clinical end point. In contrast, in the FACET study93 the addition of regular formoterol in a dose of 12 μg twice daily to either low dose (200 μg/day) or high dose (800 μg/day) budesonide reduced severe exacerbation rates by 26% in each instance. However, the reduction in severe asthma episodes for patients on low dose steroid was still best achieved by increasing the anti-inflammatory therapy (49%). Even in patients on maintenance treatment with inhaled corticosteroids and whose asthma is considered to be stable, further benefits may be achieved by adding a long acting β agonist. In the study by Taylor et al the use of salmeterol resulted in a 45% reduction in major exacerbations among patients with stable asthma, 92% of whom were already taking inhaled corticosteroids.94 Leblanc et al have reported that the benefits of long acting β agonists are greater in patients already receiving inhaled corticosteroids than in those who are not already on anti-inflammatory therapy.95 Salmeterol has also been shown to permit a reduction in maintenance dosage of inhaled corticosteroid (median reduction of 400 μg beclomethasone per day compared with 0 μg per day during the placebo period) without any apparent increase in symptoms or adverse change in lung function although, again, exacerbation rates were not reported as a study end point.96
Although the design of most of these studies may not permit final conclusions to be drawn, their results suggest that the benefits of long acting β agonists are likely to be greatest among patients already taking inhaled corticosteroids, thus raising the possibility that a positive interaction may be occurring. This seems more plausible than that long acting β agonists have intrinsic anti-inflammatory effects, clear evidence for which has not emerged. These data have formed the basis upon which the role of fixed dose combination products is now being investigated.
Conclusions
Both positive and negative interactions between corticosteroids and β agonists may occur; both have been demonstrated in vitro. On the one hand, β agonists may potentiate steroid induced repression of some cytokines while corticosteroids upregulate β receptor mRNA transcription, thus restoring β receptor function. On the other, mutual inhibition of the binding of nuclear transcription factors has been shown. These apparently divergent results may be the result of methodology; there have been important differences between tissue preparations, drug concentrations, and duration of drug exposure in the various studies. As yet we cannot conclude that these interactions—whether positive or negative—occur consistently or whether these in vitro data are relevant to the in vivo situation.
In clinical studies interactions between β agonists and corticosteroids appear to be predominantly positive. In acute severe asthma the interaction is beneficial: the administration of corticosteroid enhances the bronchodilator response to β agonist by reversing desensitisation and downregulation of β receptors. In chronic asthma there is little evidence that corticosteroid prevents β receptor downregulation. Despite this, combinations of the two drugs have been found to improve asthma control and reduce exacerbation rates. This is particularly true for long acting β agonists. The exact mechanism remains unclear. There is also the possibility that, because of a positive interaction, long acting β agonists may have “steroid enhancing” or “steroid sparing” effects. However, the use of these terms ought to be very cautiously applied: the minimum dose requirement for inhaled corticosteroids in combination therapy remains to be defined and, given that the use of long acting β agonists as monotherapy is less effective than inhaled corticosteroids alone, it is certainly not zero.
Current clinical data provide reassurance that in most patients the benefits of inhaled corticosteroids are not being compromised by β agonist use and, at least for long acting agents, are likely to be enhanced. Nevertheless, studies designed specifically to evaluate airway inflammation and bronchial hyperresponsiveness indicate that short acting β agonists may have pro-inflammatory effects and that their magnitude is not modified by treatment with inhaled corticosteroids. It remains possible that, in patients with more severe asthma or those using higher doses of drug or for longer periods of time, the pro-inflammatory actions of short acting β agonists counterbalance the benefits of inhaled corticosteroids. There are sufficient in vitro data to support this possibility. This might explain the adverse effect of β agonists on asthma control that has been observed in some studies despite concomitant anti-inflammatory treatment when a potent agent at relatively high doses has been used.97 ,98